HER2-HER3
Although the amplification and overexpression of HER2 is the oncogenic event underlying this type of cancer, it is now clear that HER3 plays a critical role in the progression of these cancers and the inactivation of constitutive HER2-HER3 signaling has turned out to be much more difficult than had been anticipated. Our efforts in this arena are currently focused on several aspects of HER2-HER3 driven tumorigenic signaling. Some are described below.
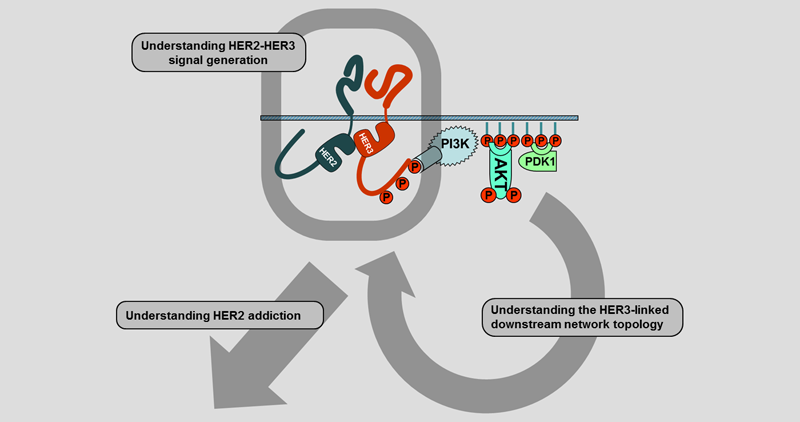
HER2-HER3 Signal generation
Work of the past decade has provided significant insight into the structural basis for the ligand-induced activation, dimerization, and transphosphorylation of HER family receptors. But many of the restraints built into the physiological mode of signaling are disrupted and overcome in the pathologic state of HER2 overexpression. Through structure-function studies spanning the realms of biochemistry to cell-based and in vivo models, we seek to understand how the massive overexpression of HER2 generates constitutive HER2-HER3 signaling, and how this differs from the normal physiological mode of regulated HER2-HER3 signaling. We are interested in whether the ligand-induced canonical extracellular domain (ECD) dimerization is necessary when HER2 is massively overexpressed. Even if ECD dimerization is not required, they can be used as targets for large biotherapeutic agents that can create steric clash and prohibit the proximation of these receptors and interfere with dimerization and activation. We have been studying these events using a variety of structure-function mutants with biochemical readouts, protein complementation assays, confocal microscopy, and in vivo tumor growth studies.

Understanding the HER3-linked downstream network topology
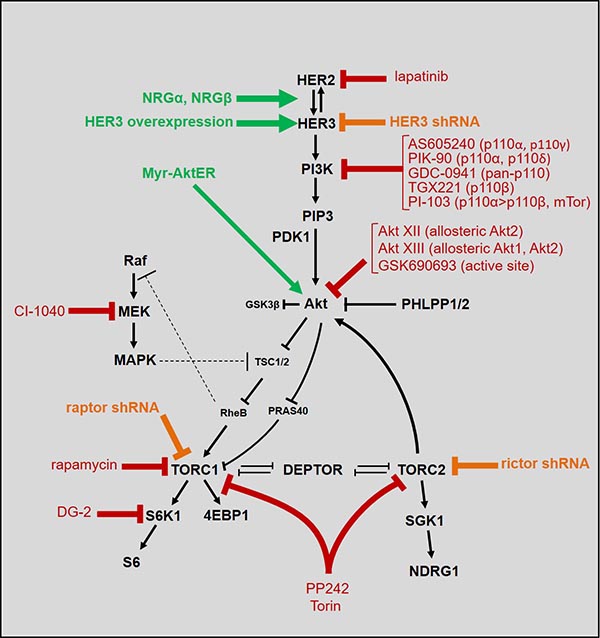
The anti-tumor activities of all classes of HER2-targeting drugs are undermined by a robust signal-buffering capacity inherent in the HER2-HER3 tumor driver and an ability to substantially and rapidly increase its signaling output to compensate for the suppressive effects of inhibitory drugs. This compensatory effect is driven by a highly competent downstream network topology linked with HER3 signaling, involving the evolutionarily conserved mTor signaling network. Using genetic and chemical genetic approaches combined with tools of computational biology, we seek to obtain a deeper understanding of this downstream network topology. In particular we are interested in how the network is altered in HER2-amplified tumor cells, and why HER3 has become an essential node in this fundamentally important eukaryotic mechanism for cellular homeostasis. We have done this using a variety of chemical, genetic, and pharmacologic techniques to stimulate or inhibit the network at various nodes and study the network response to perturbation.
Targeting HER3
Although there are a plethora of antibody and small molecule inhibitors targeting HER2, the inhibition of HER3 has been much more challenging. The HER3 kinase domain is catalytically inactive and functions in allostery not catalysis. This makes it a challenging target for the development of inhibitors. Anti-HER3 antibodies have been developed in the industry sector, with limited activities. We have been studying the HER3 kinase domain in collaboration with our structural biology collaborators, using structure-function studies to identify potential allosteric sites than can serve as targets for next generation small molecule inhibitors. We have also studied mechanisms to interfere with the cotranslational localization of HER3 that can lead to its rapid degradation.